Hoffman_nucleation_theory
ホフマン核形成理論は、1970年代と80年代にジョンD.ホフマンとその同僚によって開発された理論であり、ポリマー表面核形成の速度論と熱力学の観点からポリマーの結晶化を説明しようとしています。理論は、完全に結晶性のポリマーの表面が作成されるモデルを導入し、プロセスを説明するための表面エネルギーパラメーターを導入します。ホフマン核形成理論は、ポリマー結晶化理論の出発点であり、ホフマン-週の層状増粘におけるその基本的な役割でよく知られています。およびLauritzen–Hoffman成長理論。
コンテンツ
1 ポリマーの形態
2 核形成
3 ポリマー結晶化の熱力学
3.1 層状の肥厚(ホフマン-週プロット)
4 ポリマー結晶化の動力学
4.1 ローリッツェン-ホフマン成長理論
4.1.1 結晶化速度論の3つのレジーム
4.1.2 ポリエチレンの結晶化速度論
5 さらなるテストとアプリケーション
6 も参照してください
7 参考文献
ポリマーの形態

アモルファス領域は、結晶状態で見られるような折りたたまれた領域に秩序化するために必要なエネルギーを欠いています
ポリマーには、分子レベルでさまざまな形態が含まれており、マクロ特性が生じます。ポリマー鎖の長距離の乱れはアモルファス固体の代表であり、鎖セグメントはアモルファスと見なされます。長距離ポリマーの順序は結晶性材料に似ており、鎖セグメントは結晶性と見なされます。
ポリマーの熱特性は、ほとんどの固体材料の熱特性とは根本的に異なります。固体材料は通常、1つの融点T mを持ち、それを超えると、材料は内部の分子秩序を失い、液体になります。ポリマーは、溶融温度Tmとガラス転移温度Tgの両方を持っています。T mを超えると、ポリマー鎖は分子の秩序を失い、レプテーションまたは移動性を示します。T mより下、ただしT gより上では、ポリマー鎖は長距離移動度の一部を失い、結晶またはアモルファス領域を形成する可能性がこの温度範囲では、温度が下がると、アモルファス領域が結晶領域に遷移し、バルク材料が全体としてより結晶性になります。T g未満では、分子運動が停止し、ポリマー鎖は基本的に所定の位置で凍結します。この温度範囲では、アモルファス領域はもはや結晶領域に移行できず、ポリマーは全体として最大の結晶化度に達します。

ホフマン核形成理論は、アモルファスから結晶性ポリマーへの転移に対処し、この転移は、 TmとTgの間の温度範囲でのみ発生する可能性がアモルファスから結晶性の単一ポリマー鎖への移行は、球晶と呼ばれるさらに大きな構造のサブセットであるラメラと呼ばれる秩序ある領域を形成するために鎖のセクションを整列および折り畳むために必要なランダムな熱エネルギーに関連しています。ポリマーの結晶化は、いくつかの異なる方法で実現でき、それ自体が複雑なトピックです。
核形成
核形成とは、外面の存在の有無にかかわらず、新しい相の形成と成長です。この表面の存在は不均一な核形成をもたらしますが、存在しない場合は均一な核形成が起こります。不均一核生成は、液体または気体に浮遊している小さなダスト粒子や、 SiO 2を含むガラス表面と反応するなど、既存の核が存在する場合に発生します。ホフマン核形成のプロセスとそのローリッツェン-ホフマン成長理論への進展については、均一核形成が主な焦点です。均一な核形成は、そのような汚染物質が存在せず、あまり一般的に見られない場合に発生します。均一な核形成は、ある相から次の相へと形成される分子の小さなクラスターから始まります。クラスターが成長するにつれて、他の分子の凝縮によって凝集します。サイズは大きくなり続け、最終的に巨視的な液滴(またはシステムによっては気泡)を形成します。
核形成は、多くの場合、凝縮して液滴になる蒸気圧Pでのnモルの蒸気のギブズの自由エネルギーの変化によって数学的に説明されます。また、ポリマー結晶化における核形成障壁は、克服しなければならないエンタルピー成分とエントロピー成分の両方で構成されています。この障壁は、後で複数のレジームに関連するさまざまな長さと時間スケールで行われる選択プロセスで構成されます。この障壁は、原子核を形成するために克服するために必要な自由エネルギーです。界面の自由エネルギーであるのは、バルクから表面への核の形成です。界面の自由エネルギーは常に正の項であり、核を不安定にするように作用して、成長するポリマー鎖の継続を可能にします。核形成は好ましい反応として継続します。
ポリマー結晶化の熱力学
Lauritzen–Hoffmanプロット(右)は、(logG)+ U * / k(TT 0)が(TΔT)-1に対してプロットされた場合の3つの異なるレジームをモデル化しています。異なる温度の間で、二次核形成が成長フロントでの横方向の追加と競合する速度を説明するために使用できます。この理論は、標準的な溶融温度を含むポリマーの特性に基づいて、核形成と成長の優先度を理解するのに役立ちます。

二次核形成の3つのレジームを詳述するLauritzen–HoffmanpPlot
層状の肥厚(ホフマン-週プロット)
多くのポリマーでは、 Tcでの初期ラメラの厚さの変化はTmでの変化とほぼ同じであるため、ギブストムソン方程式でかなりうまくモデル化できます。ただし、特定の過冷却範囲(T m –T c)でのラメラの厚さは変化せず、ポリマーの多くの均一な核形成は成長フロントでの厚さの変化を意味するため、HoffmanandWeeksはより正確な表現を追求しました。この点に関して、Hoffman-Weeksプロットが作成され、方程式を介してモデル化できます。T m = T c
β + (( 1− 1 β
)。T m ∘
{ T _ { text {m}} = {T _ { text {c}} over beta} +(1- {1 over beta})T _ { text {m}} ^ { circ }}
ここで、βはL = L0βで与えられる増粘係数を表し、TcとTmはそれぞれ結晶化温度と融解温度です。
これを定数βに実験的に適用すると、 TcとTmの交点での平衡融解温度Tm°を決定できます。
ポリマー結晶化の動力学
ポリマーの結晶化プロセスは、必ずしも単純な化学反応速度式に従うとは限りません。ポリマーはさまざまな異なるレジームで結晶化する可能性があり、単純な分子とは異なり、ポリマー結晶ラメラは2つの非常に異なる表面を持っています。ポリマー結晶化速度論で最も顕著な2つの理論は、Avrami方程式とLauritzen–Hoffman成長理論です。
ローリッツェン-ホフマン成長理論
Lauritzen–Hoffman成長理論は、ポリマー結晶化の動力学を最終的に2つの速度に分解します。モデルは、成長する表面へのモノマーの追加に分解されます。この最初のステップは、一般的にポリマーの核形成に関連しています。そこから、反応速度は、ポリマーが表面で成長する速度、または横方向の成長速度になり、鎖を伸ばすポリマーへの成長速度、二次核形成速度と比較されます。これらの2つのレートは、3つの状況をもたらす可能性が
結晶化速度論の3つのレジーム

レジームIの場合、 gと呼ばれる正面の横方向の成長速度は律速段階(RDS)であり、二次核形成速度iを超えます。このg >> iの場合、単分子層は一度に1つずつ形成されるため、基板の長さがL pで厚さがbの場合、全体的な線形成長は次の式で表すことができます。G I = b I L p
{ G _ { text {I}} = biL _ { text {p}}}
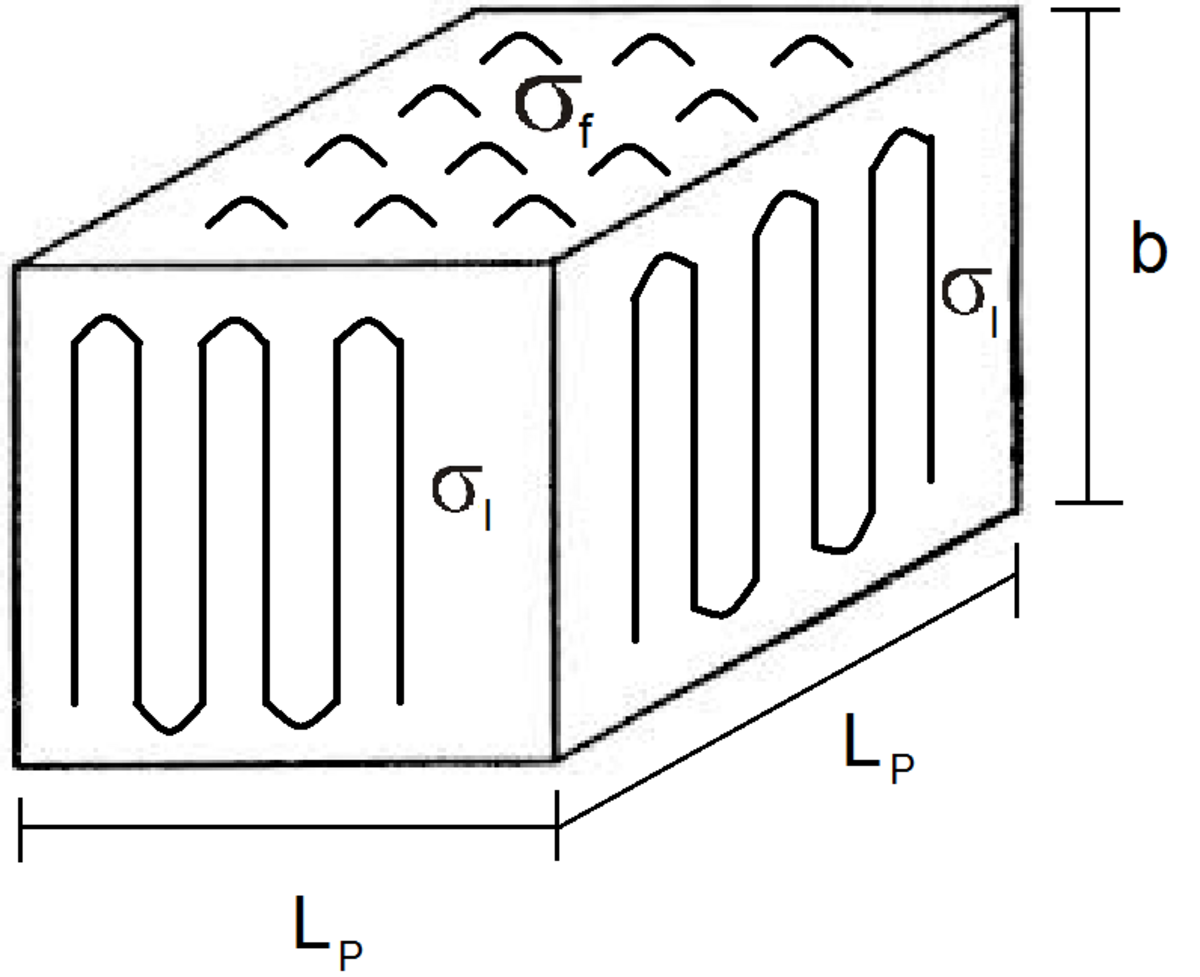
結晶性ポリマーラメラの図
具体的な核形成の速度は、次のようにさらに説明できます。
G =e −((Kg / T Δ T )。 { G _ { text {I、n}} = e ^ {-(K _ { text {g}} / T Delta T)} 、}
Kgが等しいK g = 4 b σ
l σ fT m0 Δ h
{ K _ { text {g}} = {4b sigma _ { text {l}} sigma _ { text {f}} T_ {m} ^ {0} over k Delta h}}
どこ
σlは、単位面積あたりの横方向/ラメラ表面の自由エネルギーです。
σfは、単位面積あたりの表面の折り畳み自由エネルギーです。
Tm0は平衡融解温度です
kはボルツマン定数に等しい
Δhは、標準温度での繰り返し単位あたりの融解エンタルピー(または融解潜熱)の変化に等しくなります。
これは、領域Iでは、前面に沿った横方向の核生成が溶融温度に近い温度でうまく支配することを示していますが、より極端な温度では、拡散などの他の力が核生成速度に影響を与える可能性が
レジームIIでは、横方向の成長速度は核形成速度g≤iと同等かそれよりも小さいため、最初の層が覆われる前に2次(またはそれ以上)の層が形成されます。これにより、線形成長率を次のようにモデル化できます。G II = b I g
{ G _ { text {II}} = b { sqrt {ig}}}
gとiが時間に依存しないという仮定を使用すると、新しい層が形成される速度を概算でき、レジームIIでの核形成の速度は次のように表すことができます。G II、n = e −((Kg ′ / T Δ T )。 { G _ { text {II、n}} = e ^ {-(K _ { text {g}} ^ { prime} / T Delta T)} 、}
K g ‘はレジームIのKgの約1/2に等しく、K g ′ = 2 b σ
l σ fT m0 Δ h
{ K _ { text {g}} ^ { prime} = {2b sigma _ { text {l}} sigma _ { text {f}} T_ {m} ^ {0} over k Delta h}}
最後に、LHモデルのレジームIIIは、複数の部位の核形成がi >> gを引き起こすため、横方向の成長が全体の速度に重要ではないシナリオを示しています。これは、成長率がレジームIと同じ方程式でモデル化できることを意味します。G III = b I L p= G III ∘
e− U ∗ / k(( T− T 0
)。 − ((Kg / T Δ T )。 { G _ { text {III}} = biL _ { text {p}} = G _ { text {III}} ^ { circ} e ^ {{-U ^ {*} / k(T-T_ { text {0}})}-(K _ { text {g}} / T Delta T)}}
ここで、G III °はレジームIIIの前因子であり、Lauritzen–Hoffmanプロットを適用することで実験的に決定できます。
ポリエチレンの結晶化速度論
レザの結晶化は、その鎖の層が折りたたまれて同じ方向に配向するのにかかる時間に依存します。この時間は、分子の重量と分岐とともに増加します。以下の表は、Sclair14B.1の成長率がSclair2907(20%)よりも高いことを示しています。Sclair2907は、14B.1よりも分岐が少ないです。ここで、Gcは結晶成長速度、つまり層に応じてそれ自体が注文する速度であり、tは注文にかかる時間です。
ポリマー
成長温度(°C)
G c(μm* min -1)
t(ms)
Sclair 2907(20%) 119 3.5-6.8
4.4-8.6
Sclair 14B.1 119 〜0.2
〜150
さらなるテストとアプリケーション
その後、ホフマンの原理を適用して現実と比較するために、多くの追加のテストが実行されました。行われた実験の中で、より注目すべき二次核生成テストのいくつかを以下の表で簡単に説明します。
二次核形成試験
観察された実験結果
塩化カリウム(KCl)
二次核は過冷却の程度に比例した速度で形成され(特定のレベルの攪拌を超える)、親結晶の形状に関係なく同じ量の核形成を達成します。これは、元の結晶の一次核形成よりも二次核形成の効果が大幅に大きいためです。これは、温度と形状に依存する核刺激成長実験によって証明され、二次核形成の場合、過冷却の程度と温度のみが核形成速度を変化させるのに対し、親結晶はプロセスの触媒開始剤としてのみ機能することを確認しました。
アイソタクチックポリ(ビニルシクロヘキサン)(PVCH)
PVCH結晶は、高温での広がりと横方向の成長を増加させることが実験的に示されました。これは、レジームIIIの温度に到達することは不可能でしたが、実験ポイントから3つのレジームのそれぞれで期待される動作を確認するための外挿と仮説を示しています。実験では、結晶双晶や双晶境界相互作用などの追加の成長メカニズムが従来のLH理論を変える可能性があると結論付けましたが、個々の影響をモデル化するにはさらなる研究が必要です。
酸化亜鉛(ZnO)
酸化亜鉛結晶は、ジアミンの添加や表面エッチングなどの奇妙な条件の混合下で二次核形成を受けることが証明されました。全体として、テストでは、二次結晶の形態は、基板を枯渇させ、成長を早期に妨げる能力があるため、添加するジアミンの量に応じて大きく変動する可能性があることが示されています。
も参照してください
高分子化学
高分子物理学
ポリマーの結晶化
参考文献
^ NBハンネイ(1976)。”7″。固体化学の扱い。巻 3.プレナムプレス。土井:10.1002/pol.1977.130150310。
^ チェン、スティーブン; ロッツ、バーナード(2005)。「ポリマー結晶化中の核形成障壁のエンタルピーおよびエントロピー起源:ホフマン-ローリッツェン理論およびそれ以降」。ポリマー。46(20):8662–8681。土井:10.1016/j.polymer.2005.03.125。
^ Muthukumar、M(2004)。「ポリマー結晶化における核形成」。化学物理学の進歩。128。ISBN 0-471-44528-2。
^ マランド、エルベ; Xu、Jiannong; Srinivas、Srivatsan(1998)。「ポリマー結晶の平衡融解温度の決定:線形および非線形ホフマン週の外挿」。高分子。31(23):8219–8229。Bibcode:1998MaMol..31.8219M。土井:10.1021/ma980747y。
^ Paul C. Painter、Michael M. Coleman(1997)。「8」。高分子科学の基礎入門テキスト、第2版。CRCプレス。
^ スナイダー、チャドR .; マランド、エルベ; マンスフィールド、マークL.(1996)。「Lauritzen-Hoffman二次表面核形成理論における横方向基板完成率:摩擦係数の性質」。高分子。29(23):7508–7513。Bibcode:1996MaMol..29.7508S。土井:10.1021/ma960589f。
^ スナイダー、チャドR .; マランド、エルベ(1997)。「二次表面核形成に基づくフラックス理論およびLauritzen-Hoffman結晶成長速度形式における鎖輸送の効果」。高分子。30(9):2759–2766。Bibcode:1997MaMol..30.2759S。土井:10.1021/ma961633u。
^ el Maaty、MI Abo; バセット、DC(2006年10月4日)。「溶融物からのポリエチレンの結晶化中に折り畳み表面が秩序化する時間とその分子パラメーターへの依存性について」。ポリマー。47(21):7469–7476。土井:10.1016/j.polymer.2006.08.015。
^ メリア、TP; モフィット、WP(1964)。「水溶液からの二次核形成」。工業化学化学の基礎。3(4):314–317。土井:10.1021/i160012a006。
^ アルカサル、ダニエル; ティエリー、アネット; シュルツ、パトリック; 川口明義; チェン、スティーブンZD; Lotz、Bernard(2006)。「ポリマー単結晶成長における二次核形成の横方向の広がりの程度と密度の決定」。高分子。39(26):9120–9131。Bibcode:2006MaMol..39.9120A。土井:10.1021/ma061697x。
^ Sounart、Thomas L .; 劉、6月; Voigt、James A .; フオ、メイ; Spoerke、Erik D .; マッケンジー、ボニー(2007)。「ZnOの二次核形成と成長」。混雑する。化学。Soc。129(51):15786–15793。土井:10.1021/ja071209g。”